
Research interests
Current research actitivies and interests
- Ongoing project: Currently, I am investigating cell types that are associated with complex traits, with a focus on brain related phenotypes. I systematically processed publicly available single-cell transcriptomic data from 36 human brain studies and implemented computational methods to identify disease-associated cell types
- Maintaining and developing the front-end and back-end of FUMA, a web-platform used to functionally annotate summary statistics from genome-wide association studies
- Mapping GWAS variants to genes using multi-omics quantitative trait loci
Summary of activities at Xbiome
- Developed a bioinformatics pipeline to identify tumor neoantigens from paired tumor-normal WES data from cancer patients
- Developed a bioinformatics pipeline to map tumor neoantigens to the patient’s microbiome
- Applied pipelines that I developed to analyze cancer dataset to study neoantigen diversity, crosslink bacterial antigen diversity and association to immunotherapy responses
Summary of activities at Ambry Genetics
- Processed NGS data from raw bcl files to fastq files using bcl2fastq and performed quality control of the sequencing data based on mean coverage, number of bases with different amount of coverage, or percent perfect barcode matching
- Provided bioinformatics expertise and support to wetlab scientists to troubleshoot failed sequencing runs such as analyzing contamination, on target rate, or DNA fragment sizes
- Gained knowledge and experience in NGS sequencing from different Illumina machines such as Hiseq, Nextseq, and Novaseq and Roche’s assay for ctDNA
- Acquired knowledge and experience in sequencing for confirmation of copy number variation such as Sanger sequencing or MLPA
- Developed a bioinformatics pipeline to identify somatic variants and germline variants from paired tumor-normal whole exome sequence data and from tumor-only whole exome sequence data
- Developed a bioinformatics pipeline to process RNA-seq data from raw BCL file to gene counts
Post-doc research
My post-doc research focuses on using NGS data (whole genome, whole exome, and RNA-seq) to understand human health and disease
X-inactivation

Neoepitope prediction

- Publications:
Variant calling on the sex chromosome
- Preprint: https://pmc.ncbi.nlm.nih.gov/articles/PMC12190741/
- chrX and chrY are homologous at the pseudoautosomal regions (PARs)
- Consequence: when mapping an XX sample to a default reference genome (includes both chrX and chrY), chrX reads mismap to chrY

- Solution: use a sex-complement reference genome
- Webster et al. 2019 GigaScience (I was an author on this)
- If sample is XX:
- Use a reference genome with the Y masked out
- If sample is XY:
- Use the reference genome with the PARs on the Y masked out
- Method:
- Map samples to a Default reference or a Sex-Complement reference
- Observation:
- Number of variants genotyped on chrX is increased
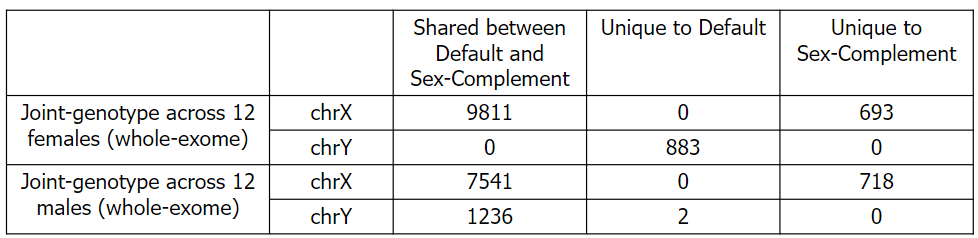
- Number of variants genotyped on chrX is increased
- Developing best practices for variant calling on the sex chromosomes:
- Mapping: Map to a Sex-Complement reference
- Genotyping:
- Joint-call within females and within males
- Male on chrX: haploid
- GATK default is diploid, which results in heterozygous sites at homologous regions with chrX
PhD Research
My research during my PhD centers around leveraging the availability of large-scale genomic data to develop methods and models for the analysis of genetic variation across species.
Natural selection
Does natural selection reduces variation at sites that are putatively neutral? Population geneticists have postulated that neutral sites could also be affected by natural selection if they are linked to those directly under selection, via genetic hitchhiking or background selection. Even though genetic diversity (DNA differences within species) at neutral sites has been shown to be reduced by linked selection, whether linked selection has also affected divergence (DNA differences between species) was still debated. I utilized genome-wide divergence data between species with different split times (i.e. human-chimp and human-mouse) to show that natural selection has also reduced divergence at linked neutral sites. I also developed a likelihood method to model different evolutionary forces, particularly linked selection, mutagenic recombination, and biased gene conversion in shaping patterns of divergence across the genome.
Published work: Phung, T.N., Huber, C.D., and Lohmueller, K.E. (2016). Determining the Effect of Natural Selection on Linked Neutral Divergence across Species. PLOS Genet 12, e1006199.
Sex-biased demography
Comparing and constrating the genetic diversity between the X chromosome and the autosomes is a tool to study whether any evolutionary process has been sex-biased and has been fruitful in providing insights into human evolutionary history. Recently, studying the genetics and the evolutionary history of dogs has captured the fascination of both scientists and the public alike. Despite documented sex-biased mating practices in both wild and domestic population of canines, current studies have not investigated whether any evolutionary process has been sex-biased. I used whole-genome sequence data from multiple populations of dogs and wolves to test whether the demography of canines has been sex-biased.
Published work: Phung, T.N., Wayne, R.K., Sayres, M.A.W., and Lohmueller, K.E. (2018). Complex patterns of sex-biased demography in canines. Proceedings of the Royal Society B 286 (1903), 20181976.
Genetic architecture
Understanding the genetic architecture of complex traits is key to unravel the genetic basis of complex traits. I leverage summary statistics from genome-wide association studies (GWAS) to develop a method to infer the mutational target size for each complex trait. I also study the extent to which selection is coupled with the variant’s effect on the trait.